Dosage du facteur de Willebrand
Généralités
- Maladie hémorragique constitutionnelle la plus fréquente.
- Prévalence estimée à 1% de la population générale.
- Transmission autosomique dominante (Types 1, 2A, 2B et 2M) ou récessive (Types 2N et 3).
- Mutations de novo exceptionnelles.
- Anomalie quantitative pour le type 1 et 3.
- Anomalie qualitative pour le type 2.
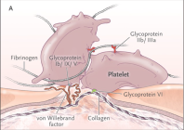
Fonctions du vWF (Facteur Von Willebrand) :
- Adhésion des plaquettes au collagène du sous-endothélium.
- Favorise l’agrégation plaquettaire.
- Transport et stabilisation du facteur VIII.
Activité du vWF dépendante de son degré de multimérisation.
Clinique variable :
- Asymptomatique.
- Hémorragies cutanéo-muqueuses (Ecchymoses, épistaxis, gingivorragies, méno-métrorragies), hémorragies post-opératoires.
- Formes sévères mimant l’hémophilie.
Variations vWF
Augmentation du vWF :
- Episode inflammatoire (maladie inflammatoire, infection…)
- Grossesse
- Oestro-progestatif
- Stress
- Exercice physique
- Sujet âgé
- Insuffisance hépatique
Diminution du vWF :
- – Groupe sanguin type O
Références
- Aillaud M.F., Facteur Willebrand, Encycl Méd Biol (Elsevier, Paris), 2003.
- Borel-Derlon A., La maladie de Willebrand, Suppl Cahier Bioforma n°20. CD-Rom Bioforma, 2004.
- Samama M.M., Elalamy l., Conardd J., Achkar A., Horellou M.H., Hémorragies et thromboses : du diagnostic au traitement, Collection « Les abrégés » Edition Masson, Paris, 2004.
- Nichols W.L., Hultin M.B., James A.H., et al. Von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia 2008; 14:171-232.
- Sadler J.E., Rodeghiero F., ISTH SSC Subcommittee on von Willebrand Factor. Provisional criteria for the diagnosis of VWD type 1. J Thromb Haemost. 2005;3(4):775-777.
- Nichols W.L.,Rick M.E., Ortel T.L, Montgomery R.R., Sadler J.E., Yawn B.P., James A.H., Hultin M.B., Manco-Johnson M.J., Weinstein M., Am J Hematol. 2009 Jun;84(6):366-370.
- Nicolas Duployez, Hématologie 2e édition, Deboeck supérieur, Louvain-la-Neuve, 2017.
- Julian Ilcheff Borissoff, M.D., Henri M.H. Spronk, Ph.D., and Hugo ten Cate, M.D., Ph.D, The Hemostatic System as a Modulator of Atherosclerosis, N Engl J Med 2011; 364:1746-1760 DOI: 10.1056/NEJMra1011670.
- Banque d’image dreamstime.

Indications du dosage
- Bilan pré-opératoire
- Syndrome hémorragique évocateur
- Allongement du TCA, diminution du facteur VIII
NB : Un TCA normal n’élimine pas le diagnostic de maladie de Willebrand.
Prescription
- Dosage de l’activité cofacteur de la Ristocétine : RCO
- Dosage de l’antigène : Ag
- Facteur VIII
- Groupe sanguin
- Plaquettes
- Fibrinogène
Exploration : RCO et Ag
- Activité cofacteur de la Riscocétine : RCO
- Etude fonctionnelle de référence : Evalue la capacité de liaison du vWF à la GpIb plaquettaire. Mesure l’agrégation de plaquettes témoins en présence du plasma du patient (contenant le vWF) et d’une concentration fixe de Ristocétine (Molécule induisant la liaison vWF-GpIb).
- Dosage antigénique du vWF:
- Dosage concentration plasmatique en vWF, indépendamment de sa fonction.
Technique immunoenzymatique avec AC anti-vWF.
- Dosage fVIII:
- Technique chronométrique. Mesure le temps de coagulation d’un mélange du plasma du patient avec un plasma déficient en facteur VIII. Le temps mesuré est exprimé en % d’activité, rapporté à une courbe de calibration établie à partir d’un plasma témoin (100% activité).
- Ratio RCO/Ag :
Si < 0.7 : anomalies qualitatives (sauf 2N)
- Ratio VIII/Ag :
- Si < 0.7 : 2N